Anomalías vasculares

2.1. Malformaciones capilares
Se manifiestan como manchas de color vinoso o rosadas, por ello también se han denominadomanchas en vino de Oporto. Hoy se sabe que las malformaciones capilares son el resultado demutaciones que se producen durante el desarrollo embrionario en genes implicados en el desarrollo de los vasos. La mayoría están causadas por mutaciones en GNAQ o GNA11 y con menor frecuencia en PIK3CA.
Estas mutaciones no se heredan, son mutaciones somáticas, lo que significa que ocurren durante el desarrollo embrionario (temprano), antes del nacimiento. Las células que adquieren la mutación crecerán y se dividirán, transmitiendo la mutación a las células que derivan de ella, pero el resto de las células serán normales. Esta situación se conoce como mosaicismo (hay células con la mutación y otras que no la tienen).
Dependiendo del momento del desarrollo en que se produzca la mutación, variará el tamaño de la mancha. Resumiendo, si la mutación ocurre muy pronto (en el periodo de embriogénesis), la mancha será de mayor tamaño. Por otro lado, si la mutación afecta a una célula que además de los vasos capilares de la piel es una célula que va a dar lugar a otras estructuras del cuerpo, pueden aparecer otras manifestaciones (como malformaciones, por ejemplo). En los casos en los que hay afectación de otros órganos, se les ha dado el nombre de diferentes síndromes. Explicamos algunos de estos síndromes a continuación:
2.1.1. Síndrome de Sturge-Weber
En este síndrome se observa una malformación capilar/mancha en “vino de Oporto” que afecta a la parte superior de la cara (frente y zona peri-orbicular) asociado a un aumento del número de capilares en las meninges (capa que recubre el cerebro y la médula espinal) y/o un aumento de capilares alrededor del ojo causando un aumento de la presión intraocular (glaucoma).
El riesgo acumulado que tiene una persona con una mancha en “vino de Oporto” en la frente para desarrollar síndrome de Sturge-Weber es de aproximadamente el 15%. También sabemos que el síndrome de Sturge-Weber es el resultado de una mutación somática en GNAQ y que no tiene factor de heredibilidad (no pasa a generaciones posteriores).
La manchas vino de Oporto ya están presentes en el momento de nacimiento, son casi siempre unilaterales (en un solo lado de la cara), aunque hay casos bilaterales. En el síndrome de Sturge-Weber la mancha afecta la frente casi en el 100% de los casos. Las manchas son planas y pueden variar en color, volviéndose más oscuras y gruesas con el tiempo
La malformación capilar de las meninges (capa de tejido conectivo especializado que cubre el cerebro y la médula espinal con fines de protección) se denomina angiomatosis leptomeníngea y solo se puede detectar realizando una resonancia magnética con contraste. Esta malformación puede que no de ningún síntoma o puede que se manifieste como si de un infarto cerebral se tratara. Los eventos similares a un derrame cerebral incluyen debilidad muscular temporal, anormalidades de la visión, migrañas y convulsiones. Las convulsiones son en su mayoría unilaterales (lo que significa que solo afectan a un lado del cerebro) y se clasifican como crisis focales. Estos episodios normalmente comienzan en las personas afectadas a la edad de 2 años. Debido a estos eventos, los pacientes con síndrome de Sturge-Weber tienen un rango de función cognitiva que varía desde inteligencia normal hasta discapacidad intelectual. Algunos de ellos también pueden presentar problemas de aprendizaje, déficit de concentración o atención e hiperactividad. Si las convulsiones no se controlan pueden conllevar un deterioro cognitivo de mayor o menor grado dependiendo de la intensidad de las convulsiones. Todo ello puede conducir a una atrofia cerebral y a que aparezcan calcificaciones en el tejido cerebral.
El glaucoma se desarrolla en pacientes con el síndrome durante su infancia o edad adulta temprana. La presión puede ser tan alta que los globos oculares pueden aparecer agrandados y abultados (exoftalmos). En aproximadamente un tercio (33%) de los pacientes con síndrome de Sturge-Weber se acumulan vasos sanguíneos anormales en el ojo (hemangiomas) en la parte posterior del globo ocular (coroides), formando un hemangioma coroideo que puede conducir a la pérdida de la visión.
Estas 3 características (mancha en vino de Oporto, angioma leptomeníngeo y glaucoma) del síndrome generalmente aparecen en el mismo lado de la cara/cuello o hemicuerpo, lo cual es importante porque si se encuentra uno se puede comenzar a investigar para encontrar los otros 2. El diagnóstico del síndrome de Sturge-Weber debe sospecharse cuando se detecta una malformación capilar en la parte superior de la cara.
La prevalencia de este síndrome es bastante baja, aunque es el síndrome de malformación capilar más frecuente, encontrándose en 1 de cada 20.000- 50.000 niños.
Defectos genéticos asociados
El síndrome de Sturge-Weber se ha relacionado con la mutación del gen GNAQ en la posición 183. Este gen es responsable de la síntesis de la proteína Gαq, que regula el desarrollo y la funcionalidad de los vasos sanguíneos.
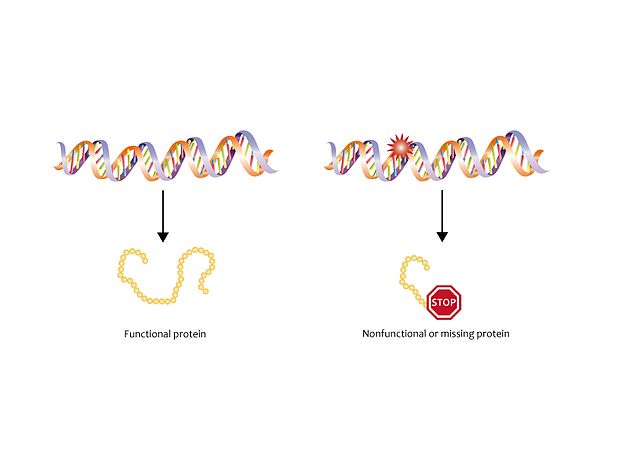
El resultado de esta mutación es la síntesis de una proteína no funcional que da como resultado la incapacidad de regular el desarrollo de nuevos vasos sanguíneos, lo que conduce a una señalización anormalmente mayor. Al aumentarse la señalización, se interrumpen las regulaciones, causando una cantidad anormal y excesiva de vasos sanguíneos y una potencial situación para la aparición de tumores vasculares. Esta interrupción de la regulación ocurre antes del nacimiento, pero los tumores vasculares aparecerán después, como se describe en el resumen de tumores vasculares y malformaciones.
La mutación no se hereda. Es una mutación somática, lo que significa que ocurre durante el desarrollo embrionario temprano. La célula que adquiere la mutación crecerá y se dividirá, transmitiendo la mutación a las células que derivan de ella. Como podemos observar después del nacimiento del niño, las células que adquieren la mutación son ciertas células del cerebro, ojos y piel involucradas en la formación de vasos sanguíneos. El resto de las células del cuerpo no presentan la mutación. Esta situación se conoce como mosaicismo, y lo mismo sucede en el síndrome de Proteus. Esta es la razón por la cual los pacientes tienen un crecimiento anormal de vasos en partes específicas de su cuerpo pero no en otras.
2.1.2. Síndrome de malformación capilar y malformación arteriovenosa
Este es un síndrome hereditario en el que aparecen manchas rosadas o marrones atenuadas, de pequeño tamaño, en la piel y de manera familiar. Las manchas van apareciendo con los años, aunque al nacer ya suele haber alguna. Además de estas manchas en la piel puede haber malformaciones arteriovenosas en las extremidades, en la columna o en el sistema nervioso central (cerebro y médula espinal).
Cuando hay malformaciones arteriovenosas acompañantes en las extremidades, esta enfermedad también se ha denominado como síndrome de Parkes-Weber. La malformacion arteriovenosa de la extremidad se acompaña a menudo de un crecimiento excesivo de la extremidad.
Defectos genéticos asociados
La causa de este síndrome se atribuye, en la mayoría de los casos, a una mutación en el gen RASA1 y en otros casos a otro gen relacionado llamado EPHB4. Esta mutación se produce en todas las células del organismo por lo que se puede detectar estudiando la sangre y por ello se pasa de padres a hijos (herencia autosómica dominante).
RASA1 codifica la proteína p120-RasGAP, que es esencial para el desarrollo normal del sistema vascular. Un RASA1 mutado codificará un p120-RasGAP deficiente o no funcional, lo que provocará malformaciones capilares y arteriovenosas.
Los casos son esporádicos, sin antecedentes en la familia, pero esta mutación en RASA1 se puede heredar y presenta un patrón autosómico dominante, lo que significa que solo se necesita 1 copia del gen (1 alelo) para adquirir el síndrome.