Vascular anomalies

2.1. Capillary malformations
These usually look like wine-coloured or pink stain and, therefore, they are also called port-wine stains. Nowadays, it is known that capillary malformations are the result of mutations that occur in the genes involved in the development of vessels during embryonic development. Most are caused due to mutations in the GNAQ or GNA11 genes and less commonly, in the PIK3CA gene.
These mutations are not inherited. They are somatic mutations, which means that they occur during early embryonic development, before birth. The cells that have the mutation will grow and divide and transmit the mutation to the cells that derive from it, however, the rest of the cells will be normal. This situation is known as mosaicism (there are cells with the mutation and others that do not have it).
.jpg){kind=link}
The size of the stain will vary depending on the development stage at the time the mutation occurs. In summary, if the mutation occurs very early (in the embryonic development period), the stain will be bigger. Conversely, if the mutation affects a cell that, as well as the capillary vessels in the skin, is a cell that is going to invade other structures in the body, other manifestations may appear (such as malformations, for example). When other organs are affected, different syndromes have been defined. Some of these syndromes are described below:
2.1.1. Sturge-Weber syndrome
In this syndrome a “port-wine” capillary malformation/stain affects the upper part of the face (forehead and periorbital region) associated with an increase in the number of capillaries in the meninges (layer covering the brain and spinal cord) and/or an increase in the number of capillaries around the eye which causes an increase in intraocular pressure (glaucoma).
The accumulated risk a person with a “port-wine” stain on the forehead has of developing Sturge-Weber syndrome is approximately 15%. We also know that Sturge-Weber syndrome is the result of a somatic mutation in the GNAQ gene which has no hereditary factor (it is not inherited by future generations).
The port-wine stains are already present at birth, they are almost always unilateral (on a single side of the face), although there are bilateral cases. In Sturge-Weber Syndrome, the stain affects the forehead in almost 100% of cases. The stains are flat and may vary in colour, and become darker and thicker over time.
The capillary malformation of the meninges (layer of specialised connective tissue that protects the brain and spinal cord) is called leptomeningeal angiomatosis and can only be detected through a magnetic resonance scan with contrast. This malformation either will not cause any symptoms or may cause a stroke. Others symptoms can betemporal muscle weakness, vision abnormalities, migraines and convulsions. Convulsions are mainly unilateral (meaning they only affect one side of the brain) and are classified as focal attacks. These episodes normally begin at the age of 2. Due to these events, the cognitive function of patients with Sturge-Weber syndrome varies from normal intelligence to intellectual disabilities. Some patients may also have learning difficulties, concentration and hyperactivity deficits. If convulsions are not controlled, they can lead to major or minor cognitive deterioration, depending on the severity. This deterioration may lead to cerebral atrophy and cause calcifications in the brain tissue.
{kind=link}
Patients with this syndrome develop glaucoma during childhood or early adulthood. Pressure can be so high that the eyeballs may seem enlarged and bulging (exophthalmos). Abnormal blood vessels accumulate in the eye (haemangiomas), in the back of the eyeball (choroid), forming a choroid haemangioma, which may lead to vision loss in approximately one third (33%) of people affected by Sturge-Weber syndrome.
These three characteristics (port-wine stain, leptomeningeal angioma, glaucoma) of the syndrome, generally appear on the same side of the face/neck or side of the body, which is important, because if one of the characteristics is found, you can then screen for the other two. The presence of Sturge-Weber syndrome should be suspected when a capillary malformation is detected on the upper part of the face.
The prevalence of this syndrome is quite low, although it is the most common capillary malformation syndrome, affecting 1 in every 20,000–50,000 children.
Associated genetic defects
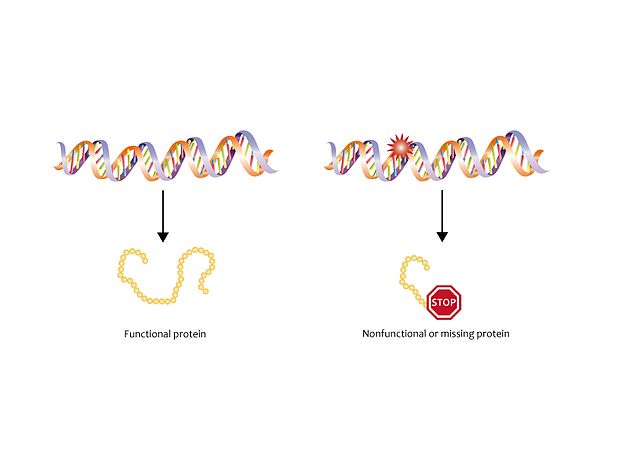
Sturge-Weber syndrome has been related to the GNAQ gene mutation in position 183. This gene is responsible for the synthesis of the Gαq protein, which regulates the development and functioning of the blood vessels.
The result of this mutation is the synthesis of a non-functional protein, which results in the inability to regulate the development of new blood vessels, leading to abnormally high signalling. When signalling increases, regulations are interrupted, causing an abnormal and excessive growth of blood vessels and a situation that promotes the formation of vascular tumours. This interruption in regulation occurs before birth, however, the vascular tumours appear later, as described in the summary on vascular tumours and malformations.
The mutation is not inherited. It is a somatic mutation, which means that it occurs during embryonic development (early), before birth. The mutated cell will grow and divide and passes on the mutation to all the derived cells. As observed after birth, the mutated cells are specifically certain cells from the brain, eyes, and skin, involved in the formation of blood vessels. The rest of the cells in the body do not have the mutation. This phenomenom is known as mosaicism, and this also happens in Proteus syndrome. This is why patients have an abnormal growth of vessels in specific parts of their bodies, but not in others.
2.1.2. Capillary malformation syndrome and arteriovenous malformation
This is a hereditary syndrome in which small faded pinkish or brown stains appear on the skin. The stains start to appear over the years, although there are usually some at birth. In addition to these skin stains, there may be arteriovenous malformations on the limbs, spinal column, or in the central nervous system (brain and spinal cord).
When there are further arteriovenous malformations on the limbs, this entity has also been named Parkes-Weber syndrome. Arteriovenous malformation of the limb is often accompanied by excessive growth of the limb.
Associated genetic defects
The cause of this syndrome is attributed, in most cases, to a mutation in the RASA1 gene and in other cases, to another related gene, called EPHB4. This mutation occurs in all the cells in the body; therefore, it can be detected by doing a blood test. It is passed from parents to children (autosomal dominant inheritance).
The p120-RasGAP protein is encoded by RASA1, which is essential for the normal development of the vascular system. A mutated RASA1 will encode a deficient or non-functioning p120-RasGAP, which will cause capillary malformations and arteriovenous malformations.
The cases are sporadic but this mutation of the RASA1 gene can be inherited and have an autosomal dominant patter, meaning that the syndrome can be inherited just by inheriting one copy of the mutated gene (1 allele).