Atrofia muscular espinal

Causas
La forma más común de AME (5q AME) afecta a las neuronas motoras inferiores. Las motoneuronas inferiores van desde la médula espinal hasta los músculos, y llevan las señales eléctricas del cerebro. Estos músculos controlan el movimiento voluntario de los brazos, las manos, la cabeza y el cuello. También controlan los músculos utilizados para respirar y tragar. El daño de las motoneuronas inferiores resulta en pérdida de masa muscular (atrofia) y la disminución de la fuerza (debilidad).
El gen SMN1
La mayoría de las personas heredan una copia del “survival motor neuron 1”, gen (SMN1) de cada padre. Los genes SMN1 contienen la información necesaria para sintetizar la proteína SMN. La proteína SMN se encarga del mantenimiento adecuado de las motoneuronas inferiores de la médula espinal.
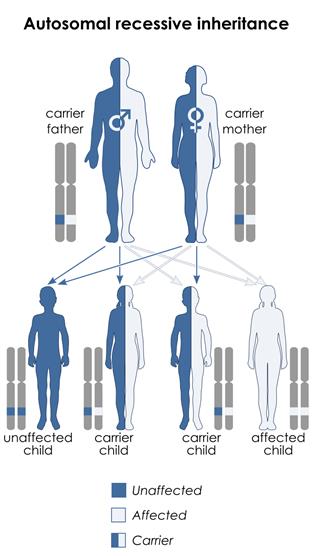
Las personas con AME 5q heredan una mutación (error) en ambas copias de su gen SMN1, uno de cada padre. Esto se conoce como herencia 'autosómica recesiva'. No son capaces de producir la proteína SMN que necesitan. Sin la proteína SMN, las neuronas motoras inferiores se deterioran. Esto limita su capacidad para transmitir las señales eléctricas del cerebro hacia los músculos afectados. Con el tiempo, esto provoca debilidad muscular, que afecta el movimiento, la respiración y la deglución.
Una mutación de novo está presente en alrededor del 2% de las personas con AME. Estas personas tienen una mutación esporádica (no heredada de sus padres), y muy probablemente es debido a una mutación en el ADN del óvulo o espermatozoide durante la concepción.
Las personas que heredan una copia defectuosa y una copia sana del gen SMN1 son portadores de la AME, lo que significa que ellos no padecen la enfermedad pero tienen el potencial para transmitirla a su descendencia. En la población general, hay una probabilidad de 1:40 de ser portador del gen defectuoso. Los portadores tienen una probabilidad de 1: 4 de transmitir la enfermedad a sus hijos, independientemente del tipo.
Los portadores de AME no muestran ningún síntoma y en consecuencia, no suelen ser conscientes de su potencial riesgo de tener un hijo afectado, a menos que tengan un hijo anterior afectado de AME.
El gen SMN2
Hay un segundo gen (el “survivor motor neuron 2” o gen SMN2), es similar al gen SMN1 y también puede producir la proteína SMN, sin embargo en una cantidad mucho más pequeña que el gen SMN1. A diferencia de la mayoría de los genes que están presentes en copias (uno heredado de la madre y otra del padre), las personas pueden tener diferentes números de copias del gen SMN2, por lo que el número de copias del gen SMN2 puede variar de persona a persona. Las personas con AME por una mutación en el gen SMN1 y que tienen más copias de SMN2, generalmente, cursarán con síntomas menos graves de AME. A pesar de esto, el gen SMN2 no puede compensar una deficiencia completa del gen SMN1, ya que carece de algunos de los bloques de construcción genéticos fundamentales del gen SMN1 para la síntesis de proteína SMN.
Enlace a la página 11-12 de la Guía de las Normas Internacionales, 2017, en Atención a la AME