Spinal muscular atrophy

Causes
The most common form of SMA (5q SMA) affects the lower motor neurons. The lower motor neurons run from the spinal cord to the muscles, and carry electrical signals from the brain. They control the voluntary movement of the arms, hands, head and neck. They also control the muscles used for breathing and swallowing. Damage to the lower motor neurons results in muscle wasting and decreased strength.
The SMN1 gene
Most people inherit one copy of the “survival motor neuron 1” (SMN1) gene from each parent. The SMN1 genes contain the information needed to make the SMN protein. The SMN protein keeps the lower motor neurons in the spinal cord healthy.
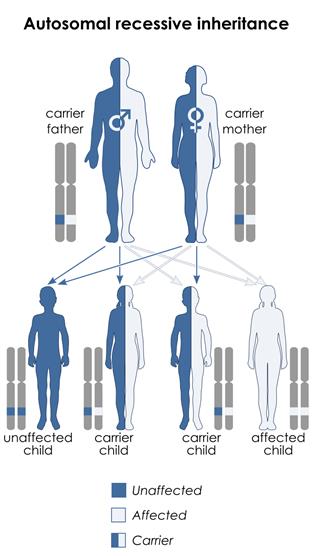
People with 5q SMA inherit a mutation (fault) in both copies of their SMN1 gene, one from each parent. This is known as ‘autosomal recessive’ inheritance. They are unable to produce the SMN protein they need. Without SMN protein, the lower motor neurons deteriorate. This restricts their ability to carry electrical signals from the brain to move the affected muscles. Over time, this causes muscle weakness, affecting movement, breathing and swallowing.
A de novo mutation is present in around 2% of people with SMA. These individuals have a new mutation (not inherited from their parents), most likely due to an error in the DNA of the egg or sperm cell during conception.
People who inherit one faulty copy and one healthy copy of the SMN1 gene are carriers of SMA, but they do not have the condition themselves. In the general population, there is a 1:40 chance of being a carrier of the faulty gene. Carriers have a 1:4 chance of their offspring being born with SMA, irrespective of type.
Carriers of SMA do not show any symptoms and therefore they are not usually aware of their potential risk of having an affected child, unless they have a previous child affected with SMA.
The SMN2 gene
There is a second gene (The “survival motor neuron 2” or SMN2 gene) which is similar to the SMN1 gene and can also produce SMN protein, however in a much smaller amount than the SMN1 gene. Unlike most genes which are present in copies (one inherited from mum and one inherited from dad), different people can have different numbers of copies of the SMN2 gene so the number of SMN2 gene copies can vary from person to person. In people with SMA due to mutations in the SMN1 gene, having more SMN2 copies is generally associated with less severe SMA symptoms. Despite this, SMN2 gene cannot fully make up for the faulty SMN1 gene as it lacks some of the key building blocks of the SMN1 gene which are essential to allow the gene to produce SMN protein.
Link to page 11-12 of the Guide to the 2017 International Standards of Care for SMA