Osteogénesis imperfecta

2.2. Genes implicados en la OI
Genes COL1A1 y COL1A2
La OI es una enfermedad genética. Aproximadamente el 90% de los individuos con OI presentan mutaciones en uno de los genes que codifican las cadenas de procolágeno I: cadena pro-α1 (gen COL1A1) y cadena pro-α2 (gen COL1A2).
Las cadenas de procolágeno I están formadas por una secuencia de aminoácidos que comprenden predominantemente un aminoácido llamado Glicina (Gly) en combinación con 2 otros aminoácidos más variables: triplete Gly-X-Y. Las mutaciones en la secuencia de bases del gen que codifica para esta proteína, producen a menudo sustituciones de esta glicina por otro aminoácido, lo que altera la cadena y la triple hélice de procolágeno I. En función de las características del aminoácido que sustituye la glicina y del punto de la cadena en la que se produce la alteración, dará lugar a una forma de OI más o menos grave.
Aparte de las sustituciones de glicina, en la OI se pueden producir otro tipo de mutaciones:
- Mutaciones que producen un cambio en el marco de lectura de los tripletes de bases (frameshift), con lo que se alterará toda la cadena de aminoácidos.
- Mutaciones en un sitio de corte y empalme del ADN (splite site).
- Cambio en una base que determina un señal de fin de la cadena de aminoácidos, con lo que la proteína no se seguirá formando a partir de este punto (nonsense).
Las formas más leves de OI se caracterizan normalmente porque la mutación implicada hace que se produzca un codón de parada prematuro de manera que la cadena de aminoácidos deja de formarse a partir de ese punto. Generalmente, el ARN obtenido es inestable y se destruye antes de que se llegue a traducir en proteína. En estos casos se produce menos cantidad de colágeno, pero es un colágeno sin alteraciones (defecto cuantitativo). En las formas más graves se sintetizan cadenas de procolágeno anómalas que se combinan con cadenas de procolágeno normales, dando lugar a una triple hélice de colágeno de estructura anómala y por lo tanto a una producción de colágeno de mala calidad (defecto cualitativo).
El 80-90% de los pacientes con OI presentan mutaciones en estos dos genes, y durante años han sido los únicos genes conocidos implicados en la OI. Los pacientes con mutaciones en estos genes tienen una variabilidad fenotípica considerable: algunos con escasas fracturas, otros con múltiples fracturas y deformidades óseas e incluso algunos casos severos con muerte intraútero o perinatal. Asimismo, pueden presentar manifestaciones clínicas extraesqueléticas diversas como escleras azules, alteraciones dentales (dentinogénesis imperfecta, malposición de los dientes…), disminución de la audición, talla baja, hiperlaxitud de las articulaciones… En los próximos capítulos se detallaran las características clínicas en detalle.
Otros genes implicados en la OI
En los últimos años se han identificado nuevos genes causantes de OI:
Genes CRTAP / P3H1 (LEPRE1) / PPIB
Las cadenas de procolágeno pro-α1 y pro-α2, una vez creadas, sufren una serie de modificaciones (llamadas modificaciones post-traduccionales) necesarias para que pueda iniciarse el ensamblaje y formación de la triple hélice. En este proceso interviene un complejo formado por 3 proteínas: CRTAP, P3H1 y CyPB. Estas proteínas están codificadas por los genes CRTAP, P3H1 (anteriormente también llamado LEPRE1) y PPIB respectivamente.
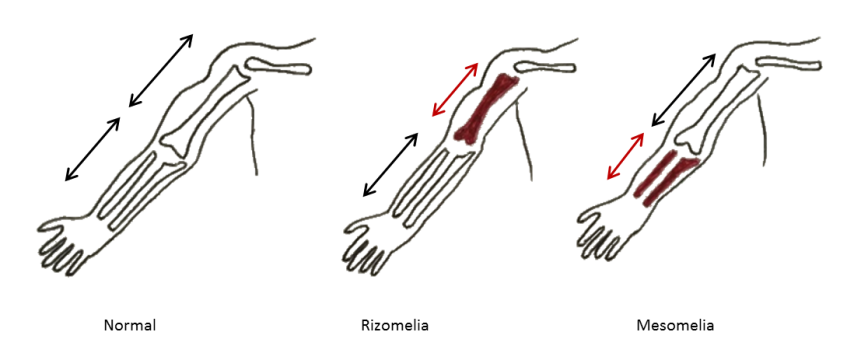
Las mutaciones en estos genes son poco frecuentes, pero producen una forma severa de OI con un fenotipo que puede ir desde casos mortales en el período perinatal hasta formas severas con huesos largos, finos y deformados y talla muy baja. Las escleras suelen ser blancas y algunos de los pacientes presentan también rizomelia, que es el acortamiento de las extremidades en su porción más proximal (el segmento hombro-codo o el segmento cadera-rodilla son más cortos) (Figura 7).
Gen FKBP10
El gen FKBP10, localizado en el cromosoma 17, codifica para una proteína llamada FKBP10 (anteriormente también llamada FKBP65), que actúa estabilizando la molécula de procolágeno y ayuda en su plegamiento en la triple hélice. Además, establece un control de calidad al proceso, evitando la secreción de moléculas con una configuración anómala.
Los pacientes con mutaciones en este gen pueden presentar 3 tipos de fenotipo:
- OI severa, de debut postnatal con fracturas y deformidades óseas progresivas y escoliosis.
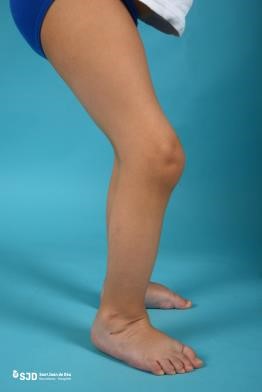
- Síndrome de Bruck: una enfermedad caracterizada por una OI severa y contracturas congénitas de las articulaciones (lo que limita su movilidad articular) (Figura 8).
- Síndrome de Kuskokwim: contracturas articulares sin fenotipo de OI severa. Hasta el momento, esta enfermedad se ha detectado únicamente en un grupo de esquimales de Alaska, y parece ser que mantienen una pequeña cantidad de proteína FKBP10 en sus células (aproximadamente un 5% de la cantidad que hay en condiciones normales) y esto les protege de padecer las características esqueléticas de la OI (aunque sí padecen las contracturas articulares).
Gen PLOD2
El gen PLOD2 codifica para la proteína LH2 ,que interviene en los cambios que ocurren tras la traducción del ADN, formando los puentes que posteriormente permitirán que las moléculas de colágeno se unan entre sí formando fibrillas estables (“crosslinking”). Se cree que LH2 mantiene una relación con la proteína FKBP10 y de hecho las mutaciones que afectan a LH2 producen también un síndrome de Bruck al igual que algunas de las mutaciones que afectan a FKBP10.
Gen SERPINH1
SERPINH1 es un gen que codifica para la proteína HSP47. En pacientes sanos, una vez formada la triple hélice, las proteínas HSP47 se unen a la cadena para hacerla más estable (evitando que se “deshaga” la hélice) y además ayuda en el transporte de la triple hélice a otra zona de la célula para continuar el proceso de formación de fibras de colágeno.
Mutaciones en este gen fueron inicialmente descritas en la raza de perros Dachshund que padecían una OI congénita. Representa una forma de OI severa pero muy poco frecuente en humanos.
Gen BMP1
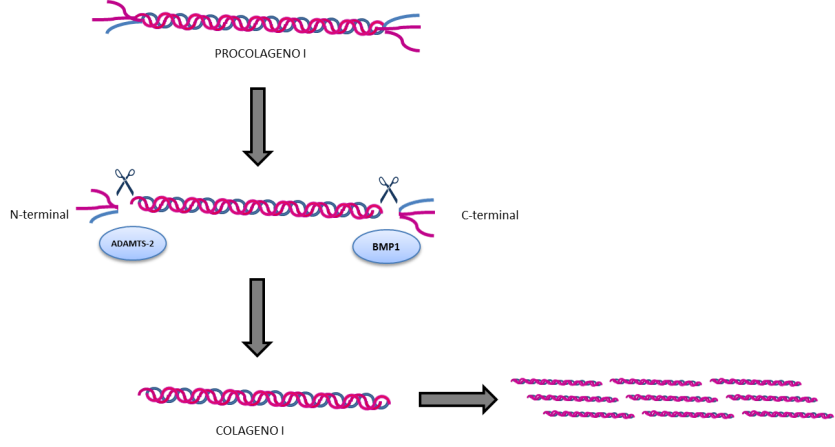
Una vez la triple hélice de colágeno está sintetizada y estabilizada, es transportada al espacio extracelular (fuera de la célula). Ahí sufre un corte de los extremos de la hélice (llamados extremos C-terminal y extremo N-terminal), que es necesario para que las moléculas de colágeno puedan ensamblarse entre sí en fibrillas y fibras de colágeno (Figura 9).
La proteína que se encarga de realizar este “corte” en el extremo C-terminal está codificada por el gen BMP1, y por ello mutaciones en este gen hacen que las fibras de colágeno no puedan organizarse correctamente y producen un fenotipo de OI que varía de formas más leves con una masa ósea elevada (más de lo que sería esperable en una OI) a formas más severas con múltiples fracturas, deformidades óseas y una baja densidad ósea.
Estudios funcionales recientes centrados en intentar dilucidar el variable grado de mineralización ósea de los diferentes pacientes descritos hasta ahora, sugieren que la molécula C-terminal que se escinde de la molécula de procolágeno podría tener también un papel en la regulación de la mineralización ósea.
Gen IFITM5
En 2012 se identificó una mutación en el gen IFITM5 que codifica para la proteína IFITM5 o BRIL. A diferencia del resto de genes implicados en la OI, la mayoría de los pacientes con mutaciones en IFITM5 identificados hasta el momento, presentan la misma mutación: c.-14C>T (hay un cambio de una citosina (C) por una timina (T) en la posición 14 del ADN). Esta mutación introduce un nuevo codón de inicio que hace que la proteína mutada que se codifica tenga 5 aminoácidos extras en uno de sus extremos. La proteína IFITM5 o BRIL se expresa en los osteoblastos, y aunque todavía no se ha podido reproducir en modelos animales, se sugiere que podría tener un efecto modulador en la mineralización del hueso.
El fenotipo de OI que traduce esta mutación tiene unas manifestaciones clínicas muy características:
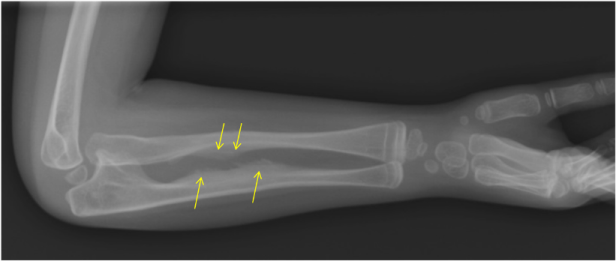
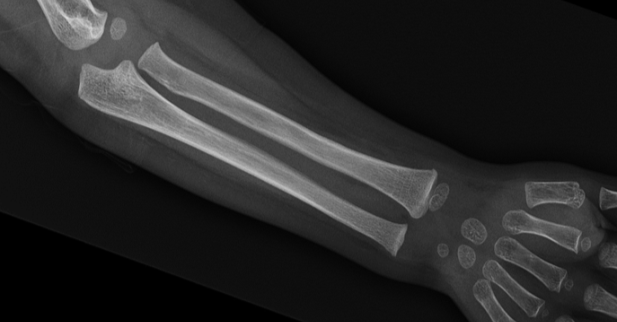
- Calcificación de la membrana interósea. Es la membrana que hay entre los huesos del cúbito y el radio del antebrazo y que en condiciones normales no está osificada y permite la movilidad normal del antebrazo. En los pacientes con OI por mutación en IFITM5 se produce una mineralización de esta membrana, con lo que se vuelve más rígida y dificulta el movimiento de giro del antebrazo (prono-supinación) (Figura 10).
- Formación de callos de fractura hipertróficos. En el proceso de reparación de una fractura el hueso es capaz de fabricar más hueso alrededor de la fractura para repararla (es lo que llamamos un callo óseo). En estos pacientes los callos son más grandes y exagerados de lo que deberían ser, lo que da la apariencia de tumores óseos alrededor de los sitios de fractura.
Gen SERPINF1
SERPINF1 es un gen localizado en el cromosoma 17 que codifica para una proteína llamada PEDF (del inglés “Pigment epithelium derived factor” o factor derivado del epitelio pigmentario). PEDF se encuentra en varios tejidos humanos como el ojo, hígado, corazón, tejido graso y hueso (donde se expresa en las células principales del hueso como los condrocitos, osteoblastos y osteoclastos).
Los pacientes con mutaciones en el gen SERPINF1 tienen niveles bajos de la proteína PEDF en sangre y presentan una forma de OI en la que cuando se analizan muestras de hueso al microscopio se observa una desorganización de las fibras de colágeno y una matriz ósea muy poco mineralizada.
La función de PEDF en el hueso y como regula su mineralización es todavía desconocida. PEDF no afecta directamente a la producción de colágeno, pero sí interviene en la unión de las moléculas de colágeno entre sí para formar fibrillas bien organizadas (por eso en estos pacientes se observa una desorganización de las fibras de colágeno). También se ha visto que PEDF afecta la función de los osteoblastos y osteoclastos.
Gen SP7
En los últimos años, se ha descrito una variante causante de OI en el gen SP7 en un paciente con una forma de OI moderada-severa. Aunque todavía queda por detallar el papel de este gen en la OI, parece ser que la proteína que codifica (llamada Osterix) no está relacionada directamente con la vía de síntesis del colágeno I. Lo que sí se conoce y se ha podido demostrar en ratones, es que es una proteína esencial en la formación del hueso, interviniendo en la diferenciación y maduración de los osteoblastos.
Gen TMEM38B
Otro de los genes descrito en pacientes con OI es TMEM38B que codifica para la proteína TRIC-B, que funciona como un canal transmembrana que interviene en la liberación de reservas de calcio intracelulares. Este gen se localiza en el cromosoma 9 y en los pocos casos con mutaciones identificadas hasta el momento, se asocia a un fenotipo de OI moderado-severo.
Gen WNT1
El gen WNT1 codifica para una proteína fundamental para la actividad formadora de hueso de los osteoblastos. Mutaciones en las dos copias del gen (recibimos una copia de cada progenitor) se han descrito en formas de OI con severidad variable, mientras que mutaciones en una sola de las dos copias del gen se han relacionado con formas de osteoporosis precoz (sin presentar el fenotipo completo de OI).
Gen CREB3L1
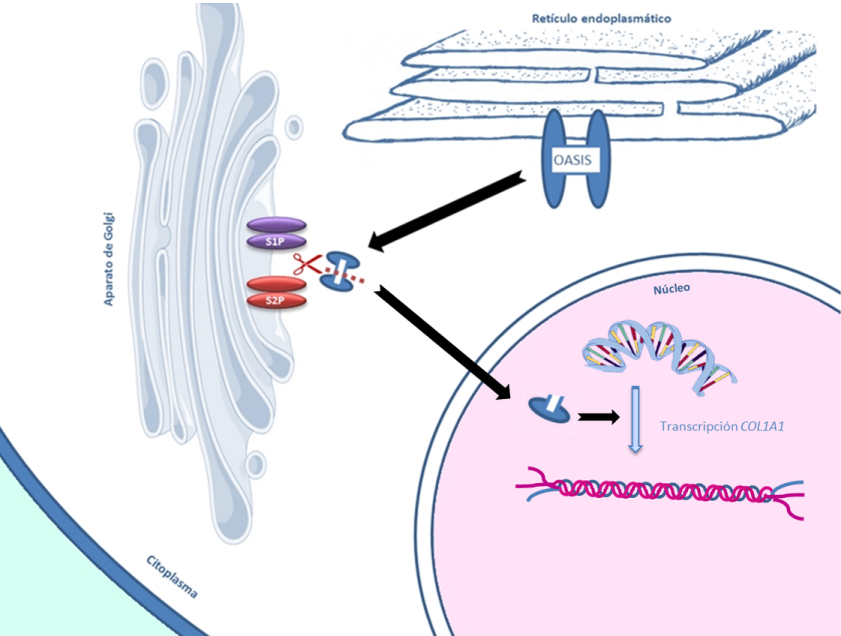
El gen CREB3L1 codifica para una proteína llamada OASIS que, en modelos de ratones, se ha visto relacionada en el proceso de transcripción de COL1A1 (proceso de lectura del ADN para formar una molécula de RNA a partir de la cual se sintetizará la cadena proteica de colágeno) (Figura 9) y en el transporte de algunas proteínas de la matriz ósea. Mutaciones en este gen se han descrito en una familia con OI muy severa.
Gen MBTPS2
MBTPS2 es un gen localizado en el cromosoma X que codifica para una proteína llamada SP2. Esta proteína, junto a la proteína S1P, interviene en un proceso llamado “proteólisis regulada intramembrana” (RIP). Este proceso tiene lugar en el aparato de Golgi y se encarga de cortar las porciones N-terminal de algunas proteínas para que puedan entrar en el núcleo de la célula y activar la transcripción de determinados genes. Una de las proteínas que requieren de SP2 para ser activadas es OASIS (mutaciones en el gen CREB3L1 que codifica esta proteína también se han relacionado con OI) (Figura 11).
Mutaciones en MBPTPS2 se habían relacionado con algunas enfermedades con afectación predominantemente de la piel, y recientemente se han descrito también en diferentes familias con miembros afectos de OI.
Gen SPARC
El gen SPARC, localizado en el cromosoma 5, codifica para una proteína llamada SPARC u osteonectina. Esta proteína se une al colágeno I y a otras proteínas de la matriz extracelular. Mutaciones en el gen SPARC se han identificado recientemente en dos niñas de diferentes familias con una forma de OI severa. Al nacimiento tenían un esqueleto normal, pero durante la infancia desarrollaron una fragilidad ósea severa con múltiples fracturas, escoliosis (desviación de la columna), hipotonía muscular (músculos más débiles) e hiperlaxitud articular.
Gen PLS3
El gen PLS3 se encuentra en el cromosoma X y codifica para una proteína llamada Plastina-3. Esta proteína se encuentra en osteoclastos, osteocitos y osteoblastos, pero su función en el hueso está todavía por determinar. Mutaciones en este gen se han encontrado en pacientes varones con osteoporosis y fracturas vertebrales y de huesos largos en la infancia. El fenotipo de estos pacientes se parece al de una OI leve, pero carece de las manifestaciones extraesqueléticas que presentan los pacientes con OI.
Gen SEC24D
Recientemente se han identificado mutaciones en el gen SEC24D en dos familias alemanas con una forma de OI con fragilidad ósea con fracturas en el período prenatal, que asociaba además unos rasgos faciales característicos: macrocefalia (cabeza grande) con una frente prominente y proptosis (ojos salidos), micrognatia (mandíbula pequeña), zonas del cráneo sin mineralizar y talla baja. Todo ello entra dentro del espectro de un síndrome muy poco frecuente llamado Cole-Carpenter.
La proteína SEC24D forma parte de un complejo de proteínas intracelulares llamado COPII que se encarga de transportar otras proteínas para que puedan realizar sus funciones. El procolágeno utiliza este complejo para ser transportado por lo que mutaciones en este gen dificultan su transporte y hacen que el procolágeno se acumule en una zona de la célula llamada retículo endoplasmático. Esto explicaría el papel de este gen en la vía de la síntesis y transporte del colágeno.
Gen P4HB
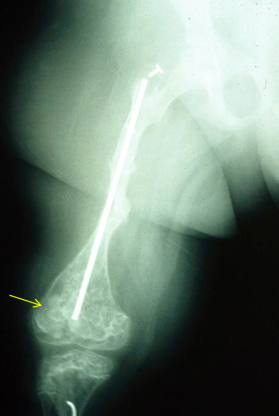
Paralelamente a los hallazgos de SEC24D, se han hallado mutaciones en P4HB en dos pacientes de dos familias diferentes, con rasgos faciales típicos del síndrome de Cole-Carpenter, que presentaban una fragilidad ósea con múltiples deformidades óseas y unos huesos largos con los extremos con aspecto de palomitas de maíz (del inglés “popcorn epiphyses”) (Figura 12).
El gen P4HB codifica para una proteína llamada PDI (del inglés “Protein Disulfide Isomerase") que participa en el ensamblaje de múltiples proteínas y también en las modificaciones post-traduccionales del procolágeno I.
Gen NBAS
El gen NBAS interviene también en el transporte de proteínas del retículo endoplasmático. Se han descrito mutaciones en este gen en diferentes enfermedades, pero recientemente se han relacionado también con pacientes con fragilidad ósea similar a la OI y otras manifestaciones como retraso del desarrollo psicomotor, inmunodeficiencia y alteración de la función del hígado.