Osteogenesis imperfecta

2.2. The genes involved in OI
COL1A1 and COL1A2 genes
OI is a genetic disease. Approximately 90% of the people with OI have mutations in one of the genes that encode the procollagen I chain: pro-α1 chain (COL1A1 gene) and pro-α2 chain (COL1A2 gene).
The procollagen I chains are made up of a sequence of amino acids predominantly comprising an amino acid called glycine (Gly) in combination with two other more variable amino acids: Gly-X-Y triplet. The mutations in the sequence of bases of the gene that encodes for this protein often produces replacement of this glycine with another amino acid, which alters the chain and the procollagen I triple helix. Depending on the characteristics of the amino acid that replaces the glycine and the point of the chain in which the alteration occurs, it will lead to a more or less severe form of OI.
In addition to glycine substitutions, there may be other types of mutations related to OI:
- Mutations that produce a change in the reading frame of base triplets (frameshift), as a result of which the entire amino acid chain is altered.
- Mutations in a DNA cut-and-paste site (splice site)
- Change in a base that determines an end signal for an amino acid chain, as a result of which the protein will not continue to form from that point (nonsense)
In the mildest forms of OI, the mutation involved causes a premature stop codon and the amino acid chain stops forming from that point on. Generally, the RNA obtained is unstable and is destroyed before it is translated into a protein. In this situation, less collagen is produced, but it is a collagen of normal quality and structure (quantitative defect). In more severe forms of OI, abnormal procollagen chains are synthesized and then combine with normal procollagen chains, giving rise to a collagen triple helix with an altered structure, resulting in the production of poor quality collagen (qualitative defect).
Eighty to ninety percent (80-90%) of patients with OI present mutations in these two genes which, for years, have been the only known genes to be involved in OI. Patients with mutations in these genes show considerable phenotypic variability: some have only few fractures, while others suffer multiple fractures and have bone deformities and even some severe cases with intrauterine or perinatal death. They can also present diverse extraskeletal clinical manifestations such as blue sclerae, dental alterations (dentinogenesis imperfecta, malpositioned teeth), hearing loss, short stature and joint hypermobility. We will examine the clinical characteristics of OI in detail in the following chapters.
Other genes involved in OI
In recent years, a number of new genes causing OI have been identified:
CRTAP / P3H1 (LEPRE1) / PPIB genes
The procollagen chains pro-α1 and pro-α2, once formed, undergo a series of modifications (called post-translational modifications) that are required to start of the assembly and formation of the triple helix. A multiprotein complex made up of three proteins (CRTAP, P3H1, and CyPB) takes part in this process. These proteins are encoded by the genes CRTAP, P3H1 (formerly called LEPRE1) and PPIB, respectively.
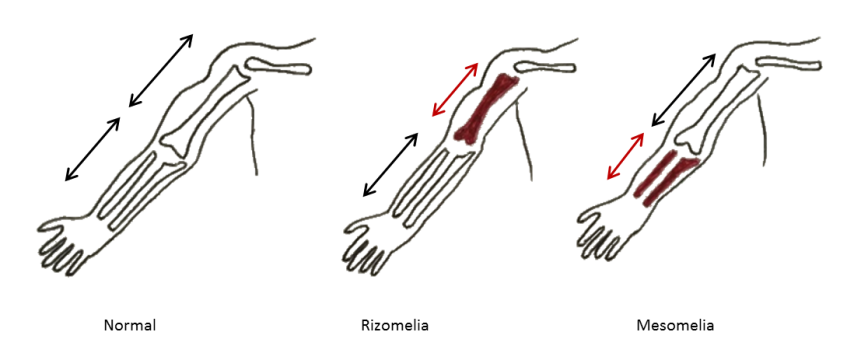
Mutations in these genes are uncommon, but when they occur they produce a severe form of OI with a phenotype that can range from lethal cases in the perinatal period to severe forms that lead to thin and deformed long bones and very short stature. The sclerae are usually white and some of the patients also present rhizomelia — a shortening of the proximal segments of the limbs (shoulder to elbow or hip to knee foreshortened) (Figure 7).
FKBP10 gene
The FKBP10 gene, located on chromosome 17, encodes a protein known as FKBP10 (formerly FKBP65), which stabilises the procollagen molecule and ensures it is folded into the triple helix. It also establishes a quality control over the process, preventing the secretion of molecules with altered configuration.
Patients with mutations in this gene may present three different phenotypes:
- Severe OI, beginning after birth with fractures and progressive bone deformities and scoliosis.
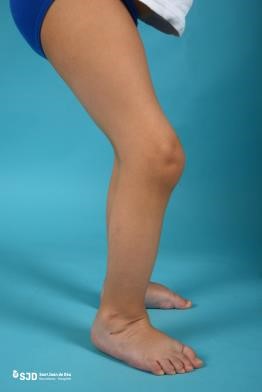
- Bruck syndrome, which is a disease characterized by severe OI and congenital joint contractures (joints with limited range of motion) (Figure 8).
- Kuskokwim syndrome, which is characterized by joint contractures without the severe OI phenotype. To date, this illness has only been detected in a group of eskimos in Alaska. It seems that they keep a small amount of the FKBP10 protein in their cells (approximately 5% of the amount found under normal conditions) and this protects them from presenting the skeletal characteristics of OI (although they do present joint contractures).
PLOD2 gene
The PLOD2 gene encodes for the protein LH2, which plays a role in post-translational changes, forming the bridges that will later allow the collagen molecules to join together to form stable fibrils (a process known as crosslinking). It is believed that LH2 is connected somehow with the protein FKBP10, and in fact the mutations that affect LH2 also produce Bruck syndrome, like some of the FKBP10 mutations.
SERPINH1 gene
SERPINH1 is a gene that encodes for the protein HSP47. In healthy patients, once the collagen triple helix is formed, HSP47 proteins bind to the chain in order to make it more stable (preventing the collapse of the helix) and it also helps in the transport of the triple helix to another area of the cell to continue the process of forming collagen fibers.
Mutations in this gene were first described in Dachshund dogs that suffered congenital OI. It represents a severe form of OI, but very uncommon in humans.
BMP1 gene
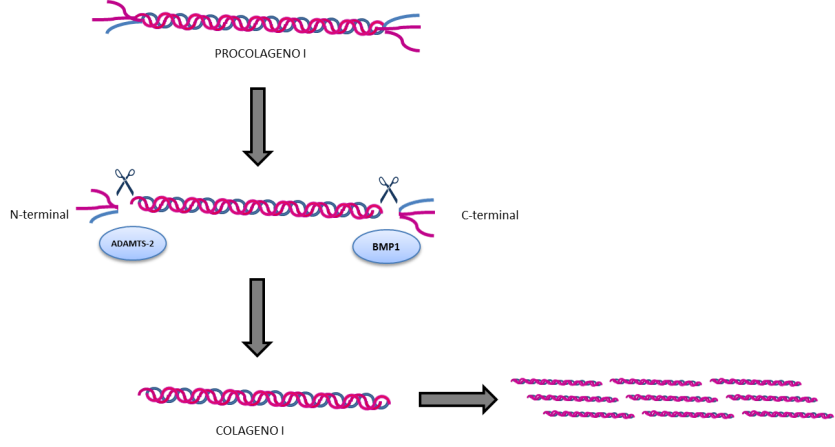
Once the collagen triple helix is synthesised and stabilised, it is transported to the extracellular space (that is, outside the cell). There, it undergoes a cleavage at the extremes of the helix (called C-terminal and N-terminal ends), required so that collagen is arranged into collagen fibrils and fibers (Figure 9).
The protein that is responsible for carrying out this cleavage in the C-terminal end is encoded by the BMP1 gene. Mutations in this gene prevent the collagen fibers from arranging themselves correctly, producing a phenotype of OI going from mild forms with increased bone mass (greater than would be expected in OI) to more severe forms with multiple fractures, bone deformities and low bone density.
Recent functional studies focused on trying to elucidate the variable degree of bone mineralisation of the patients described so far, suggest that the C-terminal molecule that is cleaved from the the procollagen molecule could also play a role in the regulation of bone mineralisation.
IFITM5 gene
In 2012 researchers identified a mutation of the IFITM5 gene, which encodes for the protein IFITM5 or BRIL. Unlike the other genes implicated in OI, the majority of patients with IFITM5 mutations identified to date have the same mutation: c.-14C>T (with the substitution of a cytosine (C) for a thymine (T) in position 14 of the DNA). This mutation introduces a new initial codon that causes the mutation of the protein, so that it has five extra amino acids at of its extremes. The protein IFITM5, or BRIL, is expressed in osteoblasts, and although it has not been reproduced yet in animal models, it appears to have a modulating effect on the mineralisation of the bone.
The phenotype of OI related to this mutation has specific clinical manifestations:
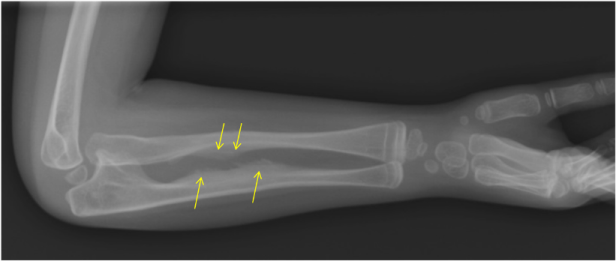
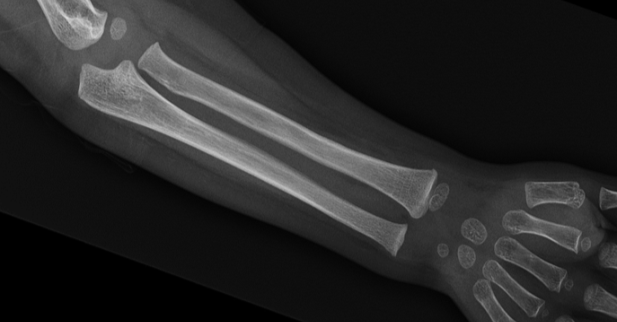
- Calcification of the interosseous membrane. This is the membrane between the ulna and the radius, which is normally not ossified, allowing the normal mobility of the forearm. In OI patients with the IFITM5 mutation, this membrane is mineralised, becoming more rigid and preventing the rotational movement of the forearm (pronosupination) (Figure 10).
- Formation of hypertrophic fracture calluses. In the process of repairing a fracture, the bone is able to produce more bone around it to mend it (what we call a bone callus). In these patients, calluses are much larger than they should be, looking like osseous tumours around the fracture site.
SERPINF1 gene
SERPINF1 is a gene located on chromosome 17 that encodes for a protein called PEDF (Pigment epithelium derived factor). PEDF is found in several human tissues such as the eye, the liver, the heart, fatty tissue, and bone (where it is expressed in the main cells of the bone, such as the chondrocytes, osteoblasts, and osteoclasts).
Patients with mutations in the SERPINF1 gene have low levels of the PEDF protein in the blood, and they present a form of OI in which, when bone samples are examined at the microscope, we see a disorganised collagen fibers and a bone matrix with very poor mineralisation.
The role of PEDF in the bone and how it regulates bone mineralisation is still unknown. PEDF does not directly affect the production of collagen, but it does intervene in the bonding of collagen molecules to form well-organised fibrils (which is why these patients present a disorganisation of collagen fibers). It has also been observed that PEDF affects the functioning of osteoblasts and osteoclasts.
SP7 gene
In recent years, a variant causing OI in theSP7 gene was described in a patient with a moderate-to-severe form of the disease. Although we still lack details concerning the role of this gene in OI, it seems that the protein that it encodes (called Osterix) is not directly related with the synthesis of collagen I. What we know, and what it has been demonstrated in rats, is that Osterix is an essential protein in bone formation, playing a role in osteoblast differentiation and maturation.
TMEM38B gene
Another gene described in patients with OI isTMEM38B, which encodes protein TRIC-B, a transmembrane channel that plays a role in liberating intracellular calcium reserves. This gene is located on chromosome 9. In the few mutations identified to date, we have seen that it is associated with a moderate-to-severe phenotype of OI.
WNT1 gene
The WNT1 gene encodes a fundamental protein for the bone-forming activity of osteoblasts. There are descriptions of mutations in the two copies of the gene (we receive one from each parent) in forms of OI with variable severity, while mutations in one of the two copies of the gene have been associated with forms of osteoporosis praecox (without presenting the complete OI phenotype).
CREB3L1 gene
The CREB3L1 gene encodes a protein called OASIS, which, in rat models, is related to COL1A1 transcription (the process of reading the DNA to form a RNA molecule, from which collagen is synthesised) and with the transport of some proteins of the bone matrix. Mutations in this gene have been detected in a family with very severe OI.
MBTPS2 gene
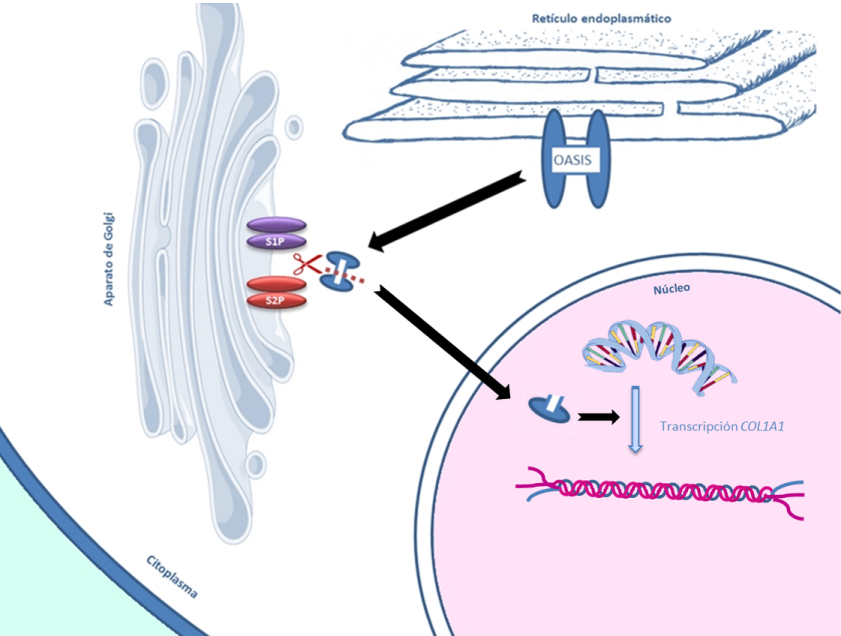
MBTPS2 is a gene located on chromosome X, which encodes a protein called SP2. This protein, along with the protein S1P, is involved in a process called ‘regulated intramembrane proteolysis’ (RIP). This process takes place in the Golgi apparatus and involves removal of the N-terminal parts of some proteins so that they can enter the cell nucleus and activate transcription of certain genes. One of the proteins that need SP2 to be activated is OASIS (mutations in the CREB3L1 gene that encode this protein have also been related with OI) (Figure 11).
Mutations in MBPTPS2 have been related with some diseases involving the skin and, recently, they have been described in families with people affected by OI.
SPARC gene
The SPARC gene, located on chromosome 5, encodes a protein known as SPARC or osteonectin. This protein bonds with collagen I and other proteins of the extracellular matrix. Mutations in the SPARC gene have been found in two young girls from different families with a severe form of OI. At birth, their skeletons were normal, but during the infancy they developed severe bone fragility with multiple fractures, scoliosis (curvature of the spine), muscular hypotonia (weak muscles), and joint hyperlaxity.
PLS3 gene
The PLS3 gene is found on chromosome X and it encodes a protein known as Plastin-3. This protein is found in osteoclasts, osteocytes and osteoblasts, but its function in the bones remains largely unknown. Mutations of this gene have been found in male patients with osteoporosis and vertebral fractures, and fractures of long bones during their infancy. The phenotype of these patients is similar to that of mild OI, but without the extraskeletal manifestations of OI.
SEC24D gene
Recently, mutations in the SEC24D gene were identified in two German families with a form of OI that includes bone fragility with prenatal fractures, and also certain facial characteristics: macrocephaly (large head) with a prominent forehead and proptosis (protruding eyes), micrognathia (small jaw), areas of the cranium without mineralization, and short stature. All of this is within the spectrum of a quite uncommon condition known as Cole-Carpenter syndrome.
SEC24D protein is part of a complex of intracellular proteins known as COPII, responsible for transporting other proteins. Procollagen is transported through this complex, so mutations in this gene prevent its transport and procollagen is accumulated in the area of the cell known as the endoplasmic reticulum. This would explain the role of this gene in the synthesis and transporting of collagen.
P4HB gene
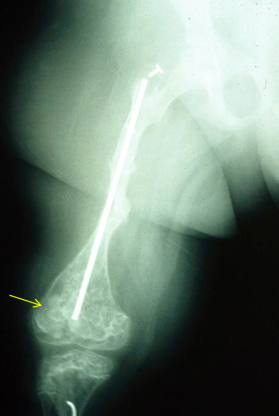
Parallel with the findings in SEC24D, mutations were also found in P4HB in two patients from two different families, with facial features typical of Cole-Carpenter syndrome, and presenting bone fragility and long bones with a popcorn-like appearance at the extremes (so-called ‘popcorn epiphysis’) (Figure 12).
P4HB encodes a protein known as PDI (protein disulfide isomerase) that participates in the assembly of multiple proteins and also in the post-translational modifications of procollagen I.
NBAS gene
The NBAS gene is also involved in the transport of proteins from the endoplasmic reticulum. Mutations in this gene have been described in several diseases, but it was recently connected as well with patients with bone fragility similar to that in OI, along with other manifestations such as delayed psychomotor development, immunodeficiency and alteration of liver function.