Osteogénesis imperfecta

3. Herencia en la OI y opciones reproductivas
Tipos de herencia
La herencia es la capacidad de transmitir a la descendencia la información que llevamos en nuestros genes. Cada gen se expresa en dos copias o alelos, una que proviene del padre y otra que proviene de la madre. La expresión final de un gen será el resultado de la interacción de los dos alelos.
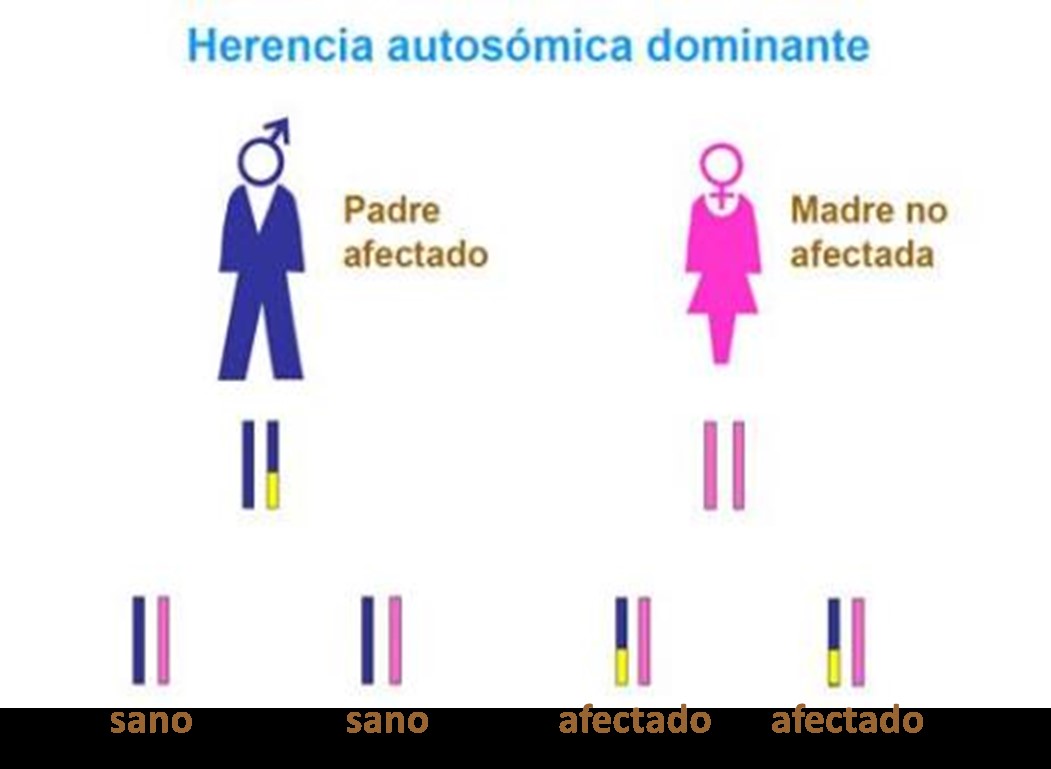
Hablamos de herencia autosómica dominante cuando una mutación en un solo alelo del gen es capaz de dar lugar a una enfermedad (Figura 13). En el caso de los hijos, si tienen una copia del gen mutado serán afectados, y si tienen las dos copias sin mutación serán sanos. En la OI, las mutaciones en los genes COL1A1, COL1A2, IFITM5 y P4HB tienen herencia autosómica dominante. Cuando la OI está causada por una mutación dominante, el gen puede ser heredado por uno de los padres o puede resultar de una mutación nueva que se ha producido en el momento de la concepción (mutación de novo). Al tratarse de un cambio estable en el ADN del niño/a, las mutaciones de novo son heredables en caso de que éste/a tenga descendencia.
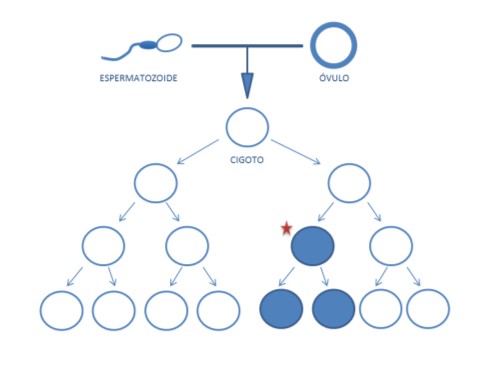
En algunos pocos casos puede ocurrir que la mutación no esté presente en todas las células del cuerpo (esto se conoce en genética como mosaicismo). Cuando un óvulo y un espermatozoide se unen, forman una célula única llamada cigoto. Esta célula se dividirá creando diferentes líneas celulares que se diferenciaran en los diferentes tejidos del organismo. El mosaicismo se produce porque aparece una mutación en este proceso de división celular (Figura 14), de manera que a partir de ese momento, únicamente las células que provengan de esta célula mutada tendrán la mutación y darán lugar a un individuo mosaico.
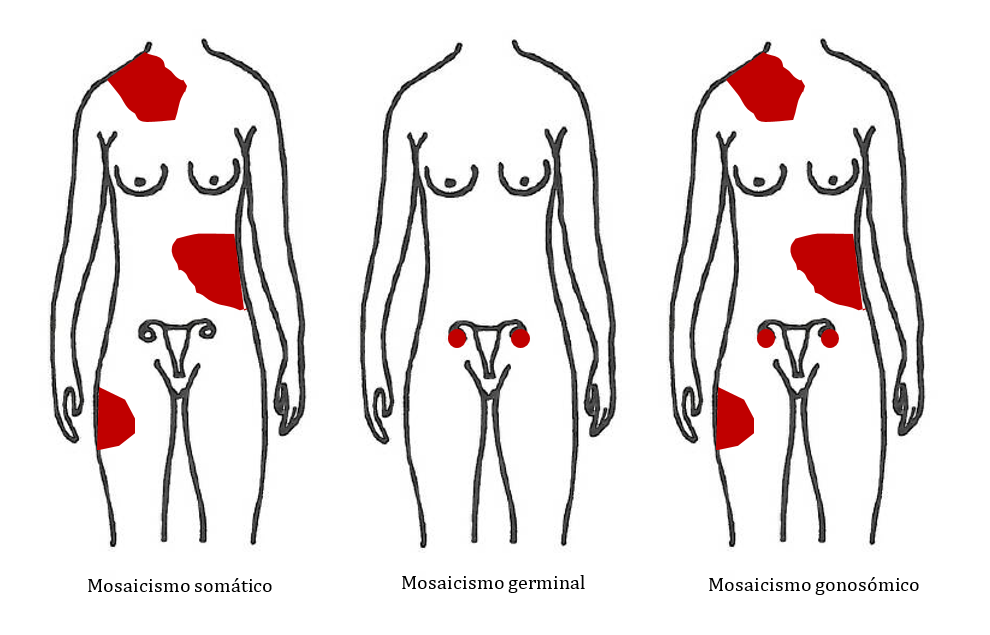
Este individuo puede no tener los síntomas de la enfermedad, pero si la mutación está presente en sus células sexuales, puede transmitir la enfermedad a la descendencia (Figura 15). En estos casos, puede parecer que se trate de una mutación de novo, porque los padres están aparentemente sanos y tienen un hijo/a afecto/a, pero cuando realizamos el estudio genético a los padres con las técnicas actuales podemos detectar la mutación en una pequeña proporción de sus células.
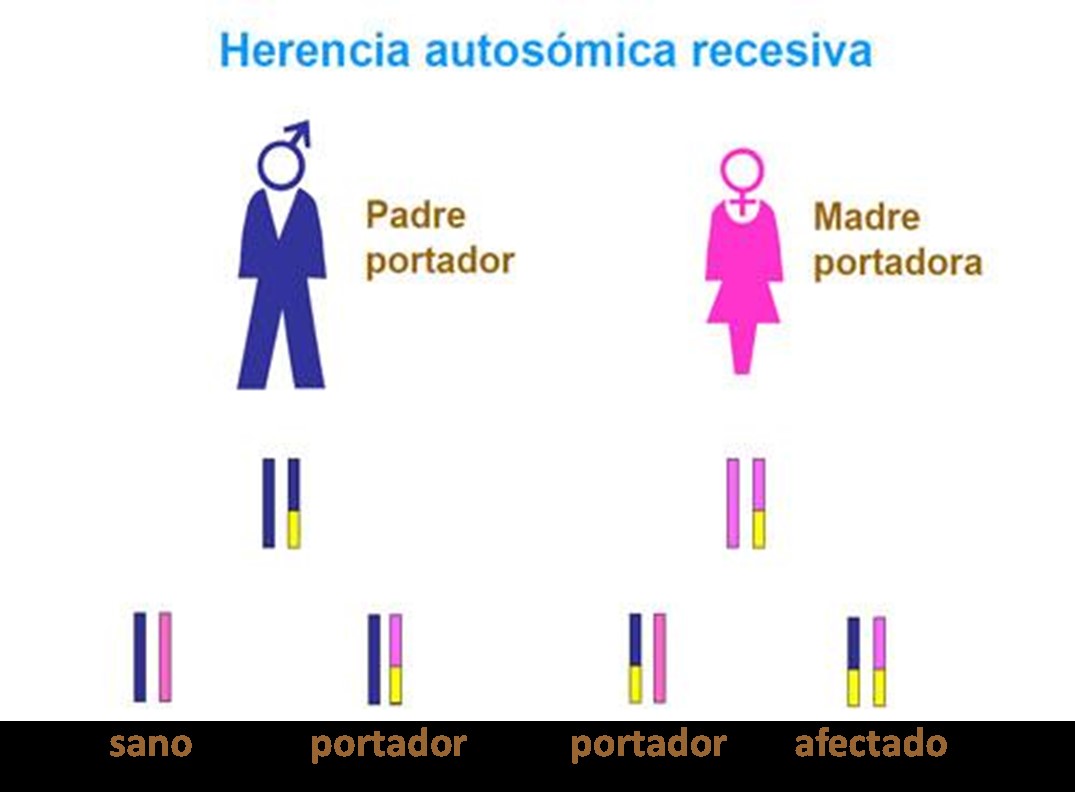
La herencia autosómica recesiva es aquella en la que se necesitan dos copias del gen mutado para que se presente la enfermedad (Figura 16). Cuando se tiene un solo alelo mutado, se es portador/a, pero este individuo no desarrolla la enfermedad. Para que un hijo tenga la enfermedad, los dos padres deben ser portadores. Los hijos que tengan dos alelos mutados serán afectados y los que tengan los dos alelos sin mutación serán sanos. En la OI, las mutaciones en los genes SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, BMP1, SP7, TMEM38B, WNT1, CREB3L1, SPARC, PLOD2, SEC24D, P4HB y NBAS presentan un patrón de herencia autosómica recesiva.
Si el paciente presenta la misma mutación en los dos alelos, se dice que la mutación está en homocigosis. Si, por el contrario, las mutaciones son diferentes, se dice que cada una de ellas está en heterocigosis. En las enfermedades de herencia recesiva el paciente debe presentar dos mutaciones, una en cada alelo del gen, para desarrollar la enfermedad. Por ello, nos podemos encontrar pacientes con 2 mutaciones iguales (mutaciones en homocigosis) o con dos mutaciones distintas en el mismo gen (ambas mutaciones en heterocigosis). En el caso de las enfermedades con herencia autosómica dominante, el paciente afecto tiene un solo alelo mutado (mutación en heterocigosis).
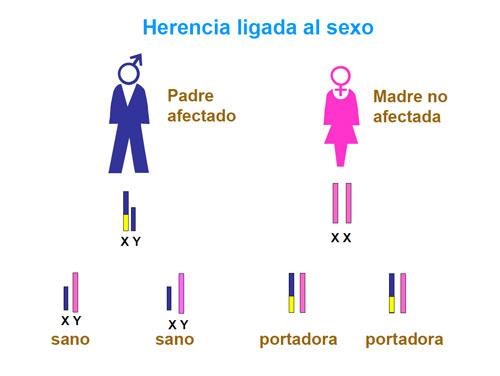
Los cromosomas X e Y determinan el sexo biológico. Los espermatozoides pueden tener un cromosoma X o uno Y, mientras que los óvulos siempre tienen un cromosoma X. Si óvulo y espermatozoide aportan cada uno un cromosoma X, el feto será niña (genotipo XX). En cambio, si el espermatozoide aporta un Y, que se une a un X aportado por el óvulo, el feto será niño (genotipo XY). Cuando el gen que transmite la enfermedad está localizado en el cromosoma X se habla de herencia ligada al sexo o herencia ligada al cromosoma X (Figura 17). En el caso de los hombres (genotipo XY), es importante recordar que sólo cuentan con una copia del cromosoma X, es decir una sola copia de ese gen. Por eso, en líneas generales, en el caso de la herencia ligada al sexo, los varones serán sanos cuando tienen el alelo del cromosoma X sin mutación o afectados cuando tienen el alelo con mutación. Las mujeres pueden ser portadoras de la enfermedad cuando tienen una copia del gen localizado en cromosoma X mutado y la otra sana. En la OI, las mutaciones en los genes MBTPS2 y PLS3 se transmiten mediante un patrón de herencia ligada al sexo.
Opciones reproductivas
La OI tiene un amplio espectro de gravedad, con formas graves que no sobreviven más allá de la etapa perinatal y otras que, aunque tienen una esperanza de vida próxima a la de los individuos sanos, implican hospitalizaciones y controles pediátricos frecuentes, tratamientos endovenosos y una carga familiar y social importante.
Por ello, ante la posibilidad de un nuevo embarazo, los padres de un niño afectado de OI pueden requerir un asesoramiento genético especializado, que les informe adecuadamente de las opciones reproductivas.
1. Diagnóstico prenatal: es el conjunto de procedimientos que permiten realizar el estudio del feto en las primeras semanas del embarazo, valorando la opción, en caso de resultar afecto, de una interrupción voluntaria del mismo. Para poder realizar un diagnóstico prenatal, es necesario conocer previamente la mutación que presenta el hijo previo afecto y haber realizado un estudio genético a los padres. Para obtener muestra del feto existen fundamentalmente dos procedimientos:
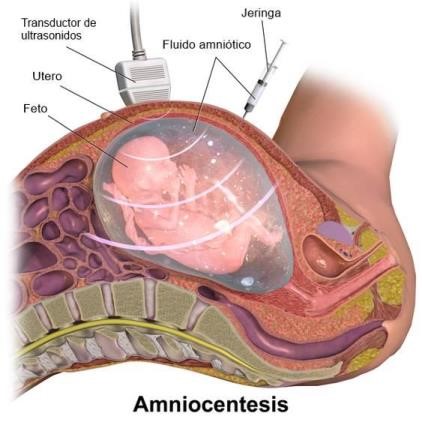
- Amniocentesis: se lleva a cabo generalmente en las semanas 16 a 18 del embarazo y se obtienen líquido amniótico por vía transabdominal (a través de la pared abdominal de la mujer embarazada). Es la técnica más habitual, la que tiene un menor riesgo de pérdida fetal (1%) y mucha fiabilidad, ya que difícilmente se contamina de muestra materna (Figura 18).
- Biopsia de corion: se lleva a cabo generalmente entre las semanas 11 a 14 del embarazo por vía transabdominal (por la pared abdominal de la mujer embarazada) o transcervical (a través de la vagina). Tiene la ventaja de que la extracción es más precoz que la amniocentesis, pero es una técnica invasiva que supone tomar una muestra de vellosidad corial a través de un catéter, por lo que tiene un mayor riesgo de pérdida fetal (2-3%).
Tras un diagnóstico prenatal mediante el estudio del feto en las primeras semanas del embarazo, en caso de resultar afecto, se puede valorar una interrupción voluntaria del mismo. Sin embargo, si se decide seguir adelante con la gestación, el tener un diagnóstico prenatal permite un seguimiento más específico del embarazo, el parto y el período postnatal.
2. Diagnóstico genético preimplantacional: permite identificar defectos genéticos en embriones fruto de una técnica in vitro, en el laboratorio, antes de ser transferidos al útero. Se estudian los embriones de forma separada y se transfieren solo aquellos que han mostrado no estar afectados genéticamente de la enfermedad que queremos evitar. Este proceso tiene una serie de pasos:
- Un ciclo de preparación de la paciente con estimulación hormonal, la inducción de la ovulación y la recogida de los oocitos mediante una punción del ovario. En cada uno de los gametos (oocitos) obtenidos de la madre, se inyecta un único espermatozoide que ha sido tomado del padre.
- A partir de entonces, se comienza a cultivar el embrión y cuando alcanza un número suficiente de células (entre los 3 y 5 días), se estudia el para detectar si presenta la mutación.
- Una vez realizado el estudio genético, se seleccionan aquellos embriones que no están afectos de la enfermedad y se transfieren al útero de la madre.
3. Donación de gametos (óvulo o espermatozoide): Otra opción para evitar la transmisión de la enfermedad es la donación de gametos de un individuo sano. Se escogerá la donación de óvulo o espermatozoide en función del progenitor que esté afecto de la enfermedad. En el caso de que se utilice un óvulo donado, lo que se realiza es una fecundación del óvulo de la donante con el esperma del padre y se procede a la transferencia del óvulo fecundado a la mujer. En el caso de donación de esperma, se realiza una inseminación artificial: se inyecta el esperma del donante en el útero de la mujer.