Distrofia miotónica

Causas
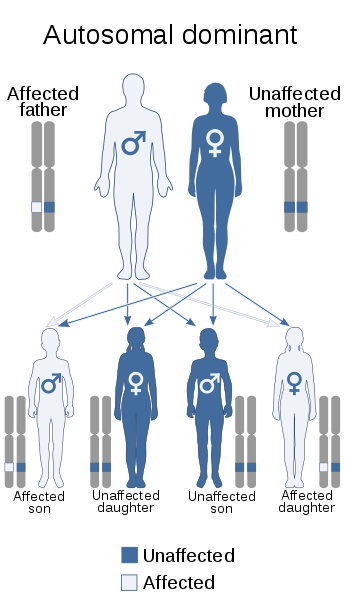
La DM1 y DM2 se heredan de manera autosómica dominante. Esto significa que un niño sólo necesita recibir una copia del gen defectuoso de uno de sus padres para heredar la enfermedad. Cualquiera de los padres puede transmitir el gen defectuoso. El padre que transmite el gen defectuoso tendrá la enfermedad, pero podría ser que no haya mostrado ningún síntoma. Es importante trazar el camino de los antecedentes familiares para minimizar su impacto en las futuras generaciones y para identificar a los miembros de la familia que podrían estar en riesgo de desarrollar los síntomas.
La causa de DM1 es una mutación genética específica en el gen DMPK. El gen DMPK se compone de bloques nucleótidos representados por las letras A, C, T y G. La tríada de C, T y G (triplete) se repite muchas veces a lo largo del gen (CTGCTGCTGCTGCTGCTGCTG...), en el gen DMPK de todos los humanos.
En una persona que no está afectada, el triplete CTG en el gen DMPK se repite entre 5 y 37 veces. En un individuo con DM1, el triplete CTG es inestable y se repite más de 37 veces. Se puede repetir miles de veces en los casos más graves.
El número de veces que se repite el triplete CTG puede ser un marcador pronóstico de la gravedad de la enfermedad. El número de repeticiones también puede determinar la edad en la que aparecerán los primeros síntomas:
- Los individuos con miles de repeticiones presentan la forma más severa, congénita (perceptible desde el nacimiento) de la enfermedad.
- Los individuos con entre 100 y miles de repeticiones pueden presentar la enfermedad de muchas maneras y puede variar la gravedad y los síntomas predominantes.
- De 50-100 / 150 repeticiones, el fenotipo de la enfermedad se considera leve o de aparición tardía. Los síntomas estarán presentes de alguna manera (por ejemplo: cataratas de aparición temprana, somnolencia diurna leve, apatía, problemas cardíacos), pero puede que no se atribuyan a unaDM1 hasta que haya un diagnóstico apropiado.
- Los individuos con 38-49 repeticiones generalmente presentan lo que llamamos una pre-mutación. Estos pacientes tienen relativamente pocos síntomas, pero tendrán una mutación frágil, que van a transmitir a sus hijos.
Es importante tener en cuenta que los síntomas más leves todavía pueden tener un impacto en la calidad de vida. Muchos de los síntomas todavía pueden ser debilitantes y requieren atención médica y apoyo.
El número de repeticiones de CTG, por lo general, aumenta de generación en generación, y esto es conocido como "Anticipación'. Un aumento en el número de repeticiones de CTG normalmente es sinónimo de una forma más severa de la enfermedad. Esto se debe a la inestabilidad de la mutación genética.
Debido a la naturaleza multisistémica de la enfermedad, no podemos suponer que la repetición es consistente, o que tiene el mismo impacto en diferentes partes del cuerpo. También es importante tener en cuenta que la repetición no es absoluta y que puede cambiar (normalmente a más) con el tiempo.
No tenemos está claro como las repeticiones de CTG interfieren con la función normal de los sistemas. Los avances científicos recientes muestran que este exceso de repeticiones de CTG conduce a la producción de ARN anormal. El ARN son copias del gen y son las responsables de ejecutar las instrucciones de los genes para la producción de proteínas esenciales. Demasiadas repeticiones de CTG en el gen DMPK afecta a la producción de ARN. El ARN adquiere una forma inusual (véase más adelante); y consecuentemente se quedan atrapados en el núcleo de la célula. Una vez atrapados, interfieren con la función de otras proteínas importantes. Estas proteínas son esenciales para diferentes funciones. Esta es probablemente la razón por la que la DM1 afecta diversos sistemas del cuerpo.

La causa de la DM2 es una mutación específica en el gen CNBP. La DM2 es una forma más leve de distrofia miotónica y los síntomas se vuelven notables en la edad adulta.
El gen CNBP contiene un complejo patrón de repetición de nucleótidos (TG)n(TCTG)n(CCTG)n. La expansión del patrón CCTG forma una zona inestable en el gen. El gen mutado codifica una versión alterada del ARN mensajero (ARNm). Las formas de ARNm anormales se agrupan y aglutinan dentro de la célula e interfieren con la producción de proteínas. Este proceso evita que las células del músculo y de otros tejidos trabajen como deberían y es la causa de DM2.
Enlace al kit de herramientas de la DM, página 23 (en inglés)