Myotonic dystrophy

Causes
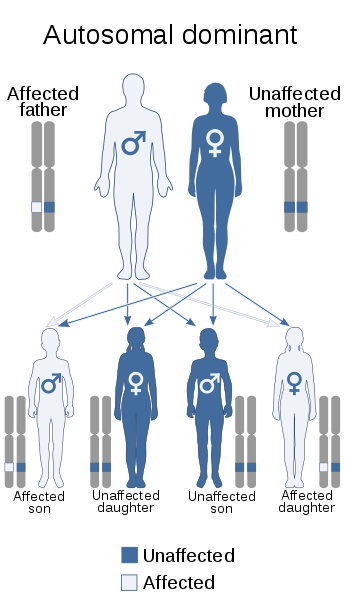
DM1 and DM2 are inherited in an autosomal dominant manner. This means that a child only needs to receive one copy of the defective gene from one of their parents to inherit the disease. Either parent can pass on the defective gene. The parent who passes on the defective gene will have the disease himself or herself, but might not show symptoms. It is important to trace the path of the disease through the family to minimise its impact on future generations, and to identify family members who might be at risk of developing symptoms.
The cause of DM1 is a specific genetic mutation in the DMPK gene. The DMPK gene consists of building blocks represented by the letters A, C, T and G. The triad of C, T and G (CTG triplet) repeats many times (e.g. CTGCTGCTGCTGCTGCTGCTG...) in everyone's DMPK gene.
In a non-affected person, the CTG triplet in the DMPK gene repeats between 5 and 37 times. In an individual with DM1, the CTG triplet is unstable and repeats more than 37 times. It can repeat thousands of times in severe cases.
The number of times this CTG triplet repeats can indicate the severity of the condition. The number of repeats can also determine the age at which the first symptoms appear:
- Individuals with thousands of repeats have the severe, congenital (noticeable at birth) form of the disease.
- Individuals with between 100 and thousands of repeats can present with the disease in a wide range of severities and predominant symptoms.
- From 50-100/150 repeats, the disease phenotype is considered mild or late onset. Symptoms will ultimately be present to some degree (e.g. early cataracts, mild daytime sleepiness, apathy, cardiac issues) but may not be attributed to DM1 until there is a proper diagnosis.
- Individuals with repeats between 38 and 49 are usually called pre-mutation. They will have relatively few symptoms, but will have a fragile mutation, which they will pass on to their children.
It is important to note that milder symptoms can still have an impact on quality of life. Many symptoms can still be debilitating and require medical care and support.
The number of CTG repeats usually increases from generation to generation, known as ‘Anticipation’. An increase in the number of CTG repeats usually results in a more severe form of the condition. This is because the change in the gene is unstable.
Due to the multi-systemic nature of the condition, we cannot assume that the repeat is consistent, or that it has the same impact on different parts of the body. It is also important to be aware that the repeat is not absolute and can change (usually increasing) over time.
We do not have a full understanding of how the CTG repeats interfere with normal function. Recent scientific advances show that too many CTG repeats lead to an abnormal RNA. RNA’s are copies of the gene and they instruct the cells to make essential proteins. Too many CTG repeats in the DMPK gene affect the production of RNA. The RNA assume an unusual shape (see below); they then become trapped in the cell nucleus. Once trapped, they interfere with the function of other important proteins. These proteins are essential for different functions. This is likely why DM1 affects many different systems in the body.

The cause of DM2 is a specific genetic mutation in the CNBP gene. DM2 is a milder form of myotonic dystrophy and symptoms become noticeable in adulthood.
The CNBP gene contains a complex repeat pattern of (TG)n(TCTG)n(CCTG)n. Expansion of the CCTG pattern forms an unstable area in the gene. The mutated gene makes an altered version of the messenger RNA (mRNA). This is a copy of the gene used for protein production. The abnormal mRNA forms clumps inside the cell and interfere with protein production. This process prevents muscle cells and other tissues from working as they should and is the cause of DM2.