Hereditary retinal dystrophies

3. X-linked retinoschisis
Under its two possible denominations (“X-linked juvenile retinoschisis” and “X-linked retinoschisis” - OMIM code 312700), it is a rare disease that only affects males. According to Orphanet, it has an estimated prevalence of 1-9/100,000 cases.
It is a retinal disease in which an abnormal separation of the retinal layers occurs and can affect central and peripheral vision. It has a slowly progressive course that produces a mild to moderate visual reduction.
It manifests as decreased vision and difficulties in reading. In severe cases, nystagmus can also be observed, but usually related to complication of the disease and not in a generalized way in patients. Fundus examination shows microcystic changes in the macular region of the retina and areas of nerve fiber layer splitting, or schisis (wheel-spoke pattern), and vitreous veils.
In severe cases, full-thickness retinal detachment occurs, leading to vision problems or blindness. In more advanced stages of the disease, vitreous hemorrhage, retinal detachment and neovascular glaucoma can be observed, which can induce severe vision loss. Female carriers rarely have vision problems.
The disease is caused by mutations in the RS1 gene, including missense, nonsense, and splice site mutations, deletions, and insertions. The RS1 gene encodes retinoschisin, an adhesive protein thought to be involved in the structural and functional integrity of the retina.
Diagnosis of X-linked retinoschisis can be clinical, based on the appearance of the fundus of the eye. An electroretinogram (ERG) shows reduced b-wave amplitude and relative preservation of the negative a-wave in the scotopic ERG (electronegative rod and mixed ERG) and in the normal photopic ERG.
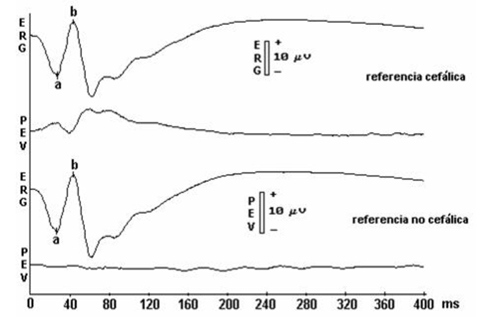
Electroretinogram, showing a and b waves
Optical coherence tomography (OCT) shows areas of schisis in the macular region. There is also a family history consistent with X-linked inheritance. Molecular genetic analysis by direct sequencing of the RS1 gene detects mutations in approximately 90% of patients. Despite this, a correlation between the type of mutation and the clinical manifestations (genotype-phenotype correlation) has not been proved to date.
Differential diagnosis includes retinitis pigmentosa and Goldmann-Favre syndrome. In X-linked retinoschisis, the disease is inherited through the mutation present on the X chromosome, so a female carrier has a 50% chance of transmitting the mutation to her offspring. Genetic testing is possible for women in the family at risk of being carriers, as well as prenatal diagnosis in high-risk pregnancies if the mutation has been identified in an affected relative.
Management of the disease includes a periodic ophthalmological examination to monitor the progression of X-linked retinoschisis. In addition, patients should be informed about possible ophthalmological complications that can be treated surgically (e.g., retinal detachment, vitreous hemorrhage, cataracts, or strabismus). Therefore, patient education and close follow-up are the only clinical alternatives for early identification and treatment of vision-threatening complications.
In X-linked retinoschisis, vision declines slowly until adolescence, and in most people it remains relatively stable through young adulthood. The disease does not progress until the forties or fifties, when there is usually a significant decline in visual acuity.
Symptoms generally begin between the ages of 10 and 30 and the progression of the disease is variable, and patients can usually maintain moderately reduced vision until late stages of the disease. Patients rarely become totally blind, however, in late stages (over 50 years of age) they can reach legal blindness, with visions lesser than 0.1.