
What causes myotonic dystrophy type 1?

Impact of neuromuscular diseases on education and working opportunities of patients and carers
Myotonic dystrophies are genetic disorders. They are systemic conditions, meaning they affect many systems in the body, not only the muscles. The genetic alteration that causes myotonic dystrophy is present at birth, but symptoms may become noticeable at almost any age. Usually, the earlier the symptoms occur, the more severe the condition will be.
There are two types of myotonic dystrophy, myotonic dystrophy type 1 (DM1, also known as Steinert’s disease) and myotonic dystrophy type 2 (DM2). Myotonic dystrophy type 1 is the most common form of myotonic dystrophy and it is often the most severe.
Myotonic dystrophies are multi-system diseases, affecting the following organs and body systems:
- Skeletal muscles (causing progressive muscle weakness and wasting)
- Energy levels and motivation
- Body movement and balance
- The heart and its electrical conduction system
- Breathing
- Swallowing
- Gut and bowels
- Vision
- The brain and cognition
- Body hormones
Myotonic dystrophies can affect these systems in different ways and to diverse degrees of severity. This is the case even among people with the same subtype and among family members.

There is currently no cure for myotonic dystrophy but there are potential new treatment options on the horizon. The general medical community does not have a good understanding of myotonic dystrophy and this can delay diagnosis and treatment.
Cause of myotonic dystrophy type 1
The cause of DM1 is a specific genetic mutation in the DMPK gene. The DMPK gene consists of building blocks represented by the letters A, C, T and G. The triad of C, T and G (CTG triplet) is repeated many times (e.g. CTGCTGCTGCTGCTGCTGCTG...) in everyone's DMPK gene.
In a non-affected person, the CTG triplet in the DMPK gene is repeated between 5 and 37 times. In an individual with DM1, the CTG triplet is unstable and the repetition appears more than 37 times. In severe cases, there are even thousands of repetitions.
The number of times this CTG triad is present in the genome has an impact on the severity of the condition. It also determines the age at which the first symptoms appear.
- Individuals with thousands of repetitions have the severe, congenital (noticeable at birth) form of the disease.
- Individuals with 35-100 repetitions have such mild symptoms that they may never receive a diagnosis.
The number of CTG replications, and therefore the severity of the condition, usually increases from generation to generation. This is because the change in the gene is unstable and the repeats can keep on expanding from one generation to the next.
We do not fully understand how the CTG repetitions interfere with normal function. Recent scientific advances show that too many of them lead to an abnormal RNA. RNAs instruct the cells to make essential proteins. Too many CTG repeats in the DMPK gene affect the production of RNA. This molecule then acquires an unusual shape (see below) and become trapped in the cell nucleus. Once trapped, they interfere with the function of other important proteins. These proteins are essential for different body functions. This is probably why DM1 affects many different systems in the body.
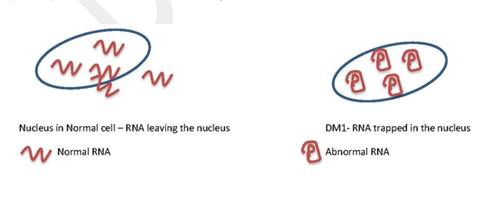
This is an excerpt from the upcoming myotonic dystrophy chapter from Share4Rare, which is currently under review. The full chapter will be available here.
Closing thoughts
Myotonic dystrophies are neuromuscular disorders that affect both children and adults. Currently, there is no cure for these pathologies and treatment is just symptomatological. Therefore, it is crucial to continue enhancing research in order to find effective treatments. Share4Rare wants to connect patients living with myotonic dystrophies to collect medical information and boost research. Will you join our global community?