Between hype and hope: the truth about gene therapy and genome editing in Duchenne muscular dystrophy

Impact of neuromuscular diseases on education and working opportunities of patients and carers
About Duchenne muscular dystrophy
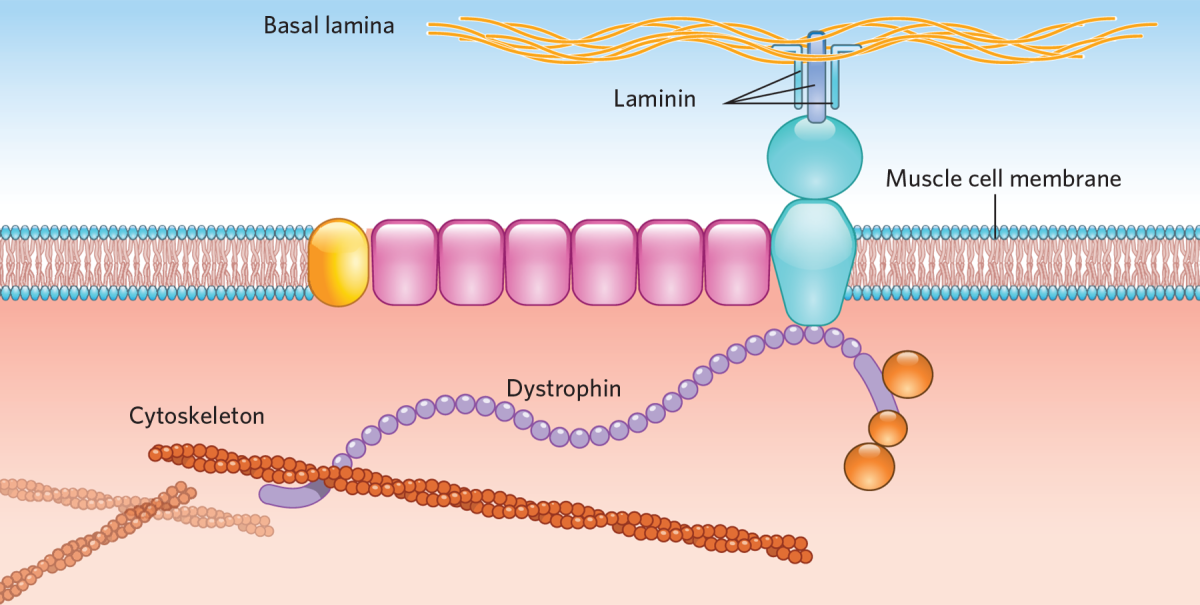
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophy affecting around 1 in 5,000 newborn boys worldwide. The main characteristic is a progressive loss of muscle function, eventually also leading to a loss of respiratory and heart function. DMD is caused by mutations in the DMD gene. This leads to the absence of the dystrophin protein, which plays an important role in the muscle cells. Without dystrophin, muscle cells easily get damaged, leading to a loss of these cells and thereby muscle function.
Treatment
Currently, the main treatment for DMD is corticosteroids, which do not cure the disease but slow down disease progression. Corticosteroids do not bring back the absent dystrophin protein. Several approaches to partly restore the dystrophin protein are currently investigated. Four medicines have been approved so far, which have moderate effects and are only suitable for specific groups of patients: the stop-codon read-through compound translarna (EMA, Europe) and the exon skipping compounds eteplirsen and golodirsen (FDA, US) and viltolarsen (MHLW, Japan).
Other therapies to replace or restore the dystrophin protein currently in different stages of development are gene therapy and gene editing. These are not a cure, but they, just like stop-codon, read-through or exon skipping, aim to enable the production of a partly functional dystrophin protein. This may slow down, but not stop disease progression, and it can certainly not reverse the loss of muscle tissue and function. There are still several hurdles that have to be overcome before these therapies could make it to the clinic.

Gene therapy in DMD
The goal of gene therapy is to add a healthy gene to the muscle cells to allow normal dystrophin production. There are several challenges for using this approach for DMD.
First of all, a large part of our body (30-40 %) consists of muscles, each containing billions of muscle cells, in turn containing billions of nuclei, in which the genes are located. A large proportion of these nuclei have to be reached before the therapy could have any effects. Delivery is a problem. Usually inactivated viral vectors are used, but the only vector that can efficiently infect muscle is the adeno-associated virus (AAV). Since AAVs are very small and the dystrophin gene is large, this does not fit. Therefore, microdystrophins are used, proteins containing only most the important parts for their function. However, it is not fully known yet how functional these microdystrophins will be in patients.
Another problem is that, due to turnover of the muscle cells — protein turnover is the continual renewal or replacement of proteins in the organism —, the genes will get lost over time and thereby dystrophin production will decrease. Unfortunately, only one-time treatment is possible, since thereafter there is immunity to the AAVs. In addition, in a large part of the population, AAV-antibodies are already pre-existing, making treatment currently not possible. Other challenges are that, at the moment, large scale manufacturing is difficult and little is known about the long-term safety of AAVs.
Currently, a few small-scale clinical trials are ongoing, revealing several problems. In the first trial very little microdystrophin production was seen, therefore, higher doses were used in subsequent trials. Since serious side effects were developed, some of these trials have temporarily been put on hold.
Genome editing
With genome editing a small part is cut out of the DNA of the mutated gene. Just like the exon skipping technique, this allows the translation of the gene to continue in order to produce a functional protein again. For this a two-component system is required: a CRISPR-guide is used, which directs Cas9 (the enzyme that cuts the DNA) to the specific location in the DNA where it should cut. As opposite to exon skipping, which occurs at the RNA level, thereby being transient (RNA is a temporary copy of the gene, produced when proteins need to be made), genome editing happens at the DNA level, so it is permanent. Therefore, this approach could have the advantage over exon skipping that only one treatment may be required.
Genome editing has so far only been tested in mouse models, not in patients, and many questions and problems still remain. In the first place, also here delivery to the muscle is an issue. Again, AAVs may be used, but since it is a two-component system both components do not fit in a single small AAV, so two AAVs are needed. This means that both an AAV containing CRISPR and an AAV containing Cas9 have to reach the same cell before it is effective. This hampers the efficiency.
Several other issues already discussed above for gene therapy play a role. Here the effect will also be lost over time, re-treatment is not possible and many patients already have pre-existing antibodies. Large-scale manufacturing is an issue too.
The same concerns on long-term safety of the use of AAVs exist. Furthermore, if the CRISPR is not specific for the targeted location, but also binds to other places in the DNA, other permanent changes in the DNA could be induced, which may be harmful. On the other hand, genome editing is, just like exon skipping, mutation-specific, meaning that it will not be suitable for all types of mutations and different CRISPR-compounds have to be developed for different mutations.
For more information on the possibilities and the misconceptions of genome editing go here.